Asymmetric Male and Female Genetic Histories among Native Americans from Eastern North America
Deborah A. Bolnick, Daniel I. Bolnick and David Glenn Smith
Molecular Biology and Evolution 2006 23(11):2161-2174
Abstract: Previous studies have investigated the human population history of eastern North America by examining mitochondrial DNA (mtDNA) variation among Native Americans, but these studies could only reconstruct maternal population history. To evaluate similarities and differences in the maternal and paternal population histories of this region, we obtained DNA samples from 605 individuals, representing 16 indigenous populations. After amplifying the amelogenin locus to identify males, we genotyped 8 binary polymorphisms and 10 microsatellites in the male-specific region of the Y chromosome. This analysis identified 6 haplogroups and 175 haplotypes. We found that sociocultural factors have played a more important role than language or geography in shaping the patterns of Y chromosome variation in eastern North America. Comparisons with previous mtDNA studies of the same samples demonstrate that male and female demographic histories differ substantially in this region. Postmarital residence patterns have strongly influenced genetic structure, with patrilocal and matrilocal populations showing different patterns of male and female gene flow. European contact also had a significant but sex-specific impact due to a high level of male-mediated European admixture. Finally, this study addresses long-standing questions about the history of Iroquoian populations by suggesting that the ancestral Iroquoian population lived in southeastern North America.
Friday, September 29, 2006
Thursday, September 28, 2006
The eight Americas of mortality
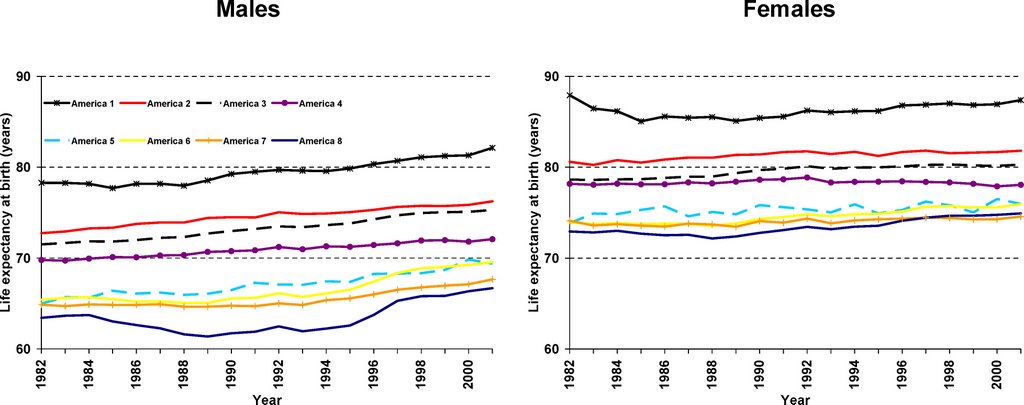
I thought the most interesting thing from this paper is how much Asians (America 1, top black line in figure below) stood out compared to the other groups as the Americans with the lowest mortality:
 Eight Americas: Investigating Mortality Disparities across Races, Counties, and Race-Counties in the United States
Eight Americas: Investigating Mortality Disparities across Races, Counties, and Race-Counties in the United States
Christopher J. L. Murray Sandeep C. Kulkarni, Catherine Michaud, Niels Tomijima, Maria T. Bulzacchelli, Terrell J. Iandiorio, Majid Ezzati
PLoS Medicine September 2006, vol. 3 Issue 9: 1513-1524
Editor's Summary
Background.
It has been recognized for a long time that the number of years that people in the United States can expect to live (“life expectancy”) varies enormously. For example, white Americans tend to live longer than black Americans, and life expectancy is much greater in some of the roughly 3,000 counties of the US than it is in others. However, there is a lack of information and understanding on how big a part is played in “health inequalities” by specific diseases and injuries, by risk factors (such as tobacco, alcohol, and obesity), and by variations in access to effective health care.
Why Was This Study Done?
The researchers wanted to find a way of dividing the people of the US into groups based on a small number of characteristics—such as location of county of residence, race, and income—that would help demonstrate the most important factors accounting for differences in life expectancy.
What Did the Researchers Do and Find?
The researchers used figures from the US Census Bureau and the National Center for Health Statistics to calculate mortality (death) rates for the years 1982–2001. They took note of the county of residence and of the race of all the people who died during that period of time. This enabled them to calculate the mortality rates for all 8,221 “race-county units” (all of the individuals of a given race in a given county). They experimented with different ways of combining the race-counties into a small and manageable number of groups. They eventually settled on the idea of there being “eight Americas,” defined on the basis of race-county, population density, income, and homicide rate. Each group contains millions or tens of millions of people. For each of the eight groups the researchers estimated life expectancy, the risk of mortality from specific diseases, the proportion of people who had health insurance, and people's routine encounters with health-care services. (The researchers also created maps of life expectancies for the US counties.) They describe their eight Americas as follows: Asians, northland low-income rural whites, Middle America, low-income whites in Appalachia and the Mississippi Valley, western Native Americans, black Middle America, low-income southern rural blacks, and high-risk urban blacks.
Many striking differences in life expectancy were found between the eight groups. For example, in 2001, the life expectancy gap between the 3.4 million high-risk urban black males and the 5.6 million Asian females was nearly 21 years. Within the sexes, the life expectancy gap between the best-off and the worst-off groups was 15.4 years for males (Asians versus high-risk urban blacks) and 12.8 years for females (Asians versus low-income rural blacks in the South). The causes of death that were mainly responsible for these variations were various chronic diseases and injury. The gaps between best-off and worst-off were similar in 2001 to what they were in 1987.
What Do These Findings Mean?
Health inequalities in the US are large and are showing no sign of reducing. Social and economic reforms would certainly help change the situation. At the same time, the public health system should also improve the way in which it deals with risk factors for chronic diseases and injuries so that groups with the highest death rates receive larger benefits.
 Eight Americas: Investigating Mortality Disparities across Races, Counties, and Race-Counties in the United States
Eight Americas: Investigating Mortality Disparities across Races, Counties, and Race-Counties in the United StatesChristopher J. L. Murray Sandeep C. Kulkarni, Catherine Michaud, Niels Tomijima, Maria T. Bulzacchelli, Terrell J. Iandiorio, Majid Ezzati
PLoS Medicine September 2006, vol. 3 Issue 9: 1513-1524
Editor's Summary
Background.
It has been recognized for a long time that the number of years that people in the United States can expect to live (“life expectancy”) varies enormously. For example, white Americans tend to live longer than black Americans, and life expectancy is much greater in some of the roughly 3,000 counties of the US than it is in others. However, there is a lack of information and understanding on how big a part is played in “health inequalities” by specific diseases and injuries, by risk factors (such as tobacco, alcohol, and obesity), and by variations in access to effective health care.
Why Was This Study Done?
The researchers wanted to find a way of dividing the people of the US into groups based on a small number of characteristics—such as location of county of residence, race, and income—that would help demonstrate the most important factors accounting for differences in life expectancy.
What Did the Researchers Do and Find?
The researchers used figures from the US Census Bureau and the National Center for Health Statistics to calculate mortality (death) rates for the years 1982–2001. They took note of the county of residence and of the race of all the people who died during that period of time. This enabled them to calculate the mortality rates for all 8,221 “race-county units” (all of the individuals of a given race in a given county). They experimented with different ways of combining the race-counties into a small and manageable number of groups. They eventually settled on the idea of there being “eight Americas,” defined on the basis of race-county, population density, income, and homicide rate. Each group contains millions or tens of millions of people. For each of the eight groups the researchers estimated life expectancy, the risk of mortality from specific diseases, the proportion of people who had health insurance, and people's routine encounters with health-care services. (The researchers also created maps of life expectancies for the US counties.) They describe their eight Americas as follows: Asians, northland low-income rural whites, Middle America, low-income whites in Appalachia and the Mississippi Valley, western Native Americans, black Middle America, low-income southern rural blacks, and high-risk urban blacks.
Many striking differences in life expectancy were found between the eight groups. For example, in 2001, the life expectancy gap between the 3.4 million high-risk urban black males and the 5.6 million Asian females was nearly 21 years. Within the sexes, the life expectancy gap between the best-off and the worst-off groups was 15.4 years for males (Asians versus high-risk urban blacks) and 12.8 years for females (Asians versus low-income rural blacks in the South). The causes of death that were mainly responsible for these variations were various chronic diseases and injury. The gaps between best-off and worst-off were similar in 2001 to what they were in 1987.
What Do These Findings Mean?
Health inequalities in the US are large and are showing no sign of reducing. Social and economic reforms would certainly help change the situation. At the same time, the public health system should also improve the way in which it deals with risk factors for chronic diseases and injuries so that groups with the highest death rates receive larger benefits.
Wednesday, September 27, 2006
Did several homo species live together at the same time?

 This is a new paper in the Journal of Human Evolution. It makes the argument that Pleistocene Homo could not have speciated into several species because we are like canids in many ways and canids have not speciated very much:
This is a new paper in the Journal of Human Evolution. It makes the argument that Pleistocene Homo could not have speciated into several species because we are like canids in many ways and canids have not speciated very much:In this paper, I use an analogy to the wolf-like canids (Canis lupus, C. rufus, and C. latrans) to ask the question, How many Homo species should there be, given their likely behavioral profile(s)? (cf. Foley, 1991). The wolf-like canids are behaviorally similar to Pleistocene hominids in three key ways: (1) they are adapted for endurance locomotion, (2) they have a diverse diet, and (3) they are socially flexible. These characteristics lead to high habitat tolerance, which appears to have made the wolf-like canids resistant to allopatric speciation. I argue that analogous habitat tolerance would have likewise made isolation and allopatric speciation among Pleistocene Homo unlikely, especially in Africa. In contrast to an earlier single-species hypothesis which posited competitive exclusion between sympatric hominid species (Wolpoff, 1971), this paper explores behavioral constraints on the process of speciation itself under conditions of temporary allopatry.
This paper cites the endurance running model of humans (D.M. Bramble and D.E. Lieberman, Endurance running and the evolution of Homo, Nature 432 (2004), pp. 345–352). I'm surprised how often that paper gets cited.
I think the paper is an important contribution, and I personally have always liked the comparison between humans and canines (cooperative hunting, highly social, cooperative child care, fission-fusion groups, etc...), and I think the issue of whether several Homo species lived together at the same time is important and fascinating, but there are still many questions that remain, as discussed by the authors in the conclusion. Here is the last paragraph:
Finally, a “species resilience” perspective on Pleistocene Homo evolution offers the potential for new comparative analyses of modern species to explore constraints on the speciation process. It has long been recognized that eurytopic mammals tend to be more widely distributed and less speciose than stenotopic ones. However, detailed quantitative analyses of mobility, dietary breadth, and genetic variation within and between eurytopic species, in conjunction with fossil evidence, are needed to resolve more precisely the determinants of relative species resilience. Especially interesting would be comparative analyses of intercontinentally distributed carnivores such as ursids, felids, and canids, as well as of widely distributed primates. Coyne and Orr (2004: 425) note that relatively little research has been devoted to factors that prevent speciation. Comparative analyses of species resilience in Homo and other eurytopes, therefore, offer the possibility of contributing both to debates about hominid taxonomy and to our understanding of mammalian speciation in general (cf. Vrba, 1992).
Species resilience in Pleistocene hominids that traveled far and ate widely: An analogy to the wolf-like canids
A. Clark Arcadia
Journal of Human Evolution Vo. 51, Issue 4 October, 2006: 383-394
Abstract: Morphological and genetic analyses have yet to resolve the question of whether more than one species of Homo existed contemporaneously in the Pleistocene. In an effort to contribute a process-related perspective to hominid phylogenetic reconstruction, this paper uses an analogy to the northern wolf-like canids (the wolves and coyotes) to ask the question, How many Homo species should there be, given their likely behavioral profile(s)? In contrast to earlier comparisons to social carnivores which sought to illuminate specific aspects of hominid behavioral ecology, this paper explores behavioral constraints on the process of speciation itself. The analogy suggests that because Pleistocene Homo probably exhibited high habitat tolerance, they would not have had the opportunity to speciate, especially in Africa. In contrast to an earlier single-species hypothesis based on competitive exclusion between sympatric hominid species, this paper explores constraints on the process of speciation under conditions of temporary allopatry.
Tuesday, September 26, 2006
Richard Dawkins book

Here's a link to Richard Dawkins' new website with a lot of info about his new book The God Delusion.
More on the genetics of human pigmentation differences
JP also has a post on this at Gene Expression.
Here's a list of genes mentioned by Shriver at al. in this paper which are implicated in pigmentation differences:
AFR - EUR
SLC24A5
MATP
OCA2
TYR
ASIP
NA- EUR
SLC24A5
MYO5A
The genetic architecture of normal variation in human pigmentation: an evolutionary perspective and model
Brian McEvoy, Sandra Beleza and Mark D. Shriver
Human Molecular Genetics 2006 15(Review Issue 2):R176-R181
Abstract: Skin pigmentation varies substantially across human populations in a manner largely coincident with ultraviolet radiation intensity. This observation suggests that natural selection in response to sunlight is a major force in accounting for pigmentation variability. We review recent progress in identifying the genes controlling this variation with a particular focus on the trait's evolutionary past and the potential role of testing for signatures of selection in aiding the discovery of functionally important genes. We have analyzed SNP data from the International HapMap project in 77 pigmentation candidate genes for such signatures. On the basis of these results and other similar work, we provide a tentative three-population model (West Africa, East Asia and North Europe) of the evolutionary–genetic architecture of human pigmentation. These results suggest a complex evolutionary history, with selection acting on different gene targets at different times and places in the human past. Some candidate genes may have been selected in the ancestral human population, others in the ‘out of Africa’ proto European-Asian population, whereas most appear to have selectively evolved solely in either Europeans or East Asians separately despite the pigmentation similarities between these two populations. Selection signatures can provide important clues to aid gene discovery. However, these should be viewed as complements, rather than replacements of, functional studies including linkage and association analyses, which can directly refine our understanding of the trait.
Here's a list of genes mentioned by Shriver at al. in this paper which are implicated in pigmentation differences:
AFR - EUR
SLC24A5
MATP
OCA2
TYR
ASIP
NA- EUR
SLC24A5
MYO5A
The genetic architecture of normal variation in human pigmentation: an evolutionary perspective and model
Brian McEvoy, Sandra Beleza and Mark D. Shriver
Human Molecular Genetics 2006 15(Review Issue 2):R176-R181
Abstract: Skin pigmentation varies substantially across human populations in a manner largely coincident with ultraviolet radiation intensity. This observation suggests that natural selection in response to sunlight is a major force in accounting for pigmentation variability. We review recent progress in identifying the genes controlling this variation with a particular focus on the trait's evolutionary past and the potential role of testing for signatures of selection in aiding the discovery of functionally important genes. We have analyzed SNP data from the International HapMap project in 77 pigmentation candidate genes for such signatures. On the basis of these results and other similar work, we provide a tentative three-population model (West Africa, East Asia and North Europe) of the evolutionary–genetic architecture of human pigmentation. These results suggest a complex evolutionary history, with selection acting on different gene targets at different times and places in the human past. Some candidate genes may have been selected in the ancestral human population, others in the ‘out of Africa’ proto European-Asian population, whereas most appear to have selectively evolved solely in either Europeans or East Asians separately despite the pigmentation similarities between these two populations. Selection signatures can provide important clues to aid gene discovery. However, these should be viewed as complements, rather than replacements of, functional studies including linkage and association analyses, which can directly refine our understanding of the trait.
Thursday, September 21, 2006
Young Australopithecus skull and partial skeleton found

This has been all over the news. I thought I'd just mention the fact that this juvenile had a large hyoid bone, and that according to the shape of the scapula, humerus, tibia and femur, the legs are very different from chimps and gorillas, but the arms are still "ape-like". The authors discuss that the arm structure can be interpreted as just a lagging phylogenetic remnant or as evidence of the importance of arborealism in their "locomotor repertoire".
This skeleton was actually found five or six years ago, if I remember correctly. I also found it interesting that they cited the Nature paper about endurance running in Homo (not in a particulary interesting context, however).
A juvenile early hominin skeleton from Dikika, Ethiopia
Zeresenay Alemseged, Fred Spoor, William H. Kimbel, René Bobe, Denis Geraads, Denné Reed and Jonathan G. Wynn
Nature 443, 296-301(21 September 2006)
Abstract: Understanding changes in ontogenetic development is central to the study of human evolution. With the exception of Neanderthals, the growth patterns of fossil hominins have not been studied comprehensively because the fossil record currently lacks specimens that document both cranial and postcranial development at young ontogenetic stages. Here we describe a well-preserved 3.3-million-year-old juvenile partial skeleton of Australopithecus afarensis discovered in the Dikika research area of Ethiopia. The skull of the approximately three-year-old presumed female shows that most features diagnostic of the species are evident even at this early stage of development. The find includes many previously unknown skeletal elements from the Pliocene hominin record, including a hyoid bone that has a typical African ape morphology. The foot and other evidence from the lower limb provide clear evidence for bipedal locomotion, but the gorilla-like scapula and long and curved manual phalanges raise new questions about the importance of arboreal behaviour in the A. afarensis locomotor repertoire.
Wednesday, September 20, 2006
RNA gene, HAR1, involved in cortical development, changed in humans
The authors suggest that we should be looking at the 98.5% of the genome that is non-coding to look for signatures of selection in humans. They find several regions that they call HAR (human accelerated region). They find HAR1 in particular that is very changed in humans compared to other primates and mammals. They also provide some functional data as to how this gene is expressed in the neocortex during the very early stages of development.
They also find that the substitutions in HAR1 ( and most of all HAR) are all A or T to C or G, so called weak-to-strong changes, that "strengthen RNA helices" and perhaps "increase gene stability and increased expression levels".
Here the Wikipedia entry on RNA genes.
An RNA gene expressed during cortical development evolved rapidly in humans
Katherine S. Pollard, Sofie R. Salama, Nelle Lambert, Marie-Alexandra Lambot, Sandra Coppens, Jakob S. Pedersen, Sol Katzman, Bryan King, Courtney Onodera, Adam Siepel, Andrew D. Kern, Colette Dehay, Haller Igel, Manuel Ares, Jr, Pierre Vanderhaeghen and David Haussler
Nature 443, 167-172(14 September 2006)
Abstract: The developmental and evolutionary mechanisms behind the emergence of human-specific brain features remain largely unknown. However, the recent ability to compare our genome to that of our closest relative, the chimpanzee, provides new avenues to link genetic and phenotypic changes in the evolution of the human brain. We devised a ranking of regions in the human genome that show significant evolutionary acceleration. Here we report that the most dramatic of these 'human accelerated regions', HAR1, is part of a novel RNA gene (HAR1F) that is expressed specifically in Cajal–Retzius neurons in the developing human neocortex from 7 to 19 gestational weeks, a crucial period for cortical neuron specification and migration. HAR1F is co-expressed with reelin, a product of Cajal–Retzius neurons that is of fundamental importance in specifying the six-layer structure of the human cortex. HAR1 and the other human accelerated regions provide new candidates in the search for uniquely human biology.
They also find that the substitutions in HAR1 ( and most of all HAR) are all A or T to C or G, so called weak-to-strong changes, that "strengthen RNA helices" and perhaps "increase gene stability and increased expression levels".
Here the Wikipedia entry on RNA genes.
An RNA gene expressed during cortical development evolved rapidly in humans
Katherine S. Pollard, Sofie R. Salama, Nelle Lambert, Marie-Alexandra Lambot, Sandra Coppens, Jakob S. Pedersen, Sol Katzman, Bryan King, Courtney Onodera, Adam Siepel, Andrew D. Kern, Colette Dehay, Haller Igel, Manuel Ares, Jr, Pierre Vanderhaeghen and David Haussler
Nature 443, 167-172(14 September 2006)
Abstract: The developmental and evolutionary mechanisms behind the emergence of human-specific brain features remain largely unknown. However, the recent ability to compare our genome to that of our closest relative, the chimpanzee, provides new avenues to link genetic and phenotypic changes in the evolution of the human brain. We devised a ranking of regions in the human genome that show significant evolutionary acceleration. Here we report that the most dramatic of these 'human accelerated regions', HAR1, is part of a novel RNA gene (HAR1F) that is expressed specifically in Cajal–Retzius neurons in the developing human neocortex from 7 to 19 gestational weeks, a crucial period for cortical neuron specification and migration. HAR1F is co-expressed with reelin, a product of Cajal–Retzius neurons that is of fundamental importance in specifying the six-layer structure of the human cortex. HAR1 and the other human accelerated regions provide new candidates in the search for uniquely human biology.
Tuesday, September 19, 2006
How much sun is optimal?
Here is a report of a paper about a finding that incidence of kidney cancer in a sample of 175 countries is positively correlated with increased distance from the equator. Here's the link to the story (not the paper), and an excerpt from the story:
The researchers created a graph with a vertical axis for renal cancer incidence rates, and a horizontal axis for latitude. The latitudes range from -90 for the southern hemisphere, to zero for the equator, to +90 for the northern hemisphere. They then plotted incidence rates for 175 countries according to latitude. The resulting chart was a parabolic curve that looks like a smile (see accompanying images).
The researchers created a graph with a vertical axis for renal cancer incidence rates, and a horizontal axis for latitude. The latitudes range from -90 for the southern hemisphere, to zero for the equator, to +90 for the northern hemisphere. They then plotted incidence rates for 175 countries according to latitude. The resulting chart was a parabolic curve that looks like a smile (see accompanying images).
"The plot points created a curve roughly resembling a smile, with countries with high incidence rates at the left and right, and those with low incidence rates in the center, just a few degrees from the equator," said Garland. "Countries with the highest cancer rates were places like New Zealand and Uruguay in the southern hemisphere and Iceland and the Czech Republic in the northern hemisphere. Clustered at the bottom of the curve with lowest incidence rates were Guam, Indonesia and other equatorial countries on most continents, including many varied equatorial cultures."
In addition to UVB, the researchers analyzed cloud cover and intake of calories from animal sources for their association to kidney cancer. The scientists were able to determine the contributions of each independently. After accounting for cloud cover and intake of animal protein, UVB exposure still showed a significant independent association with incidence rates.
"Because the distinctive "smiley" parabolic curve is present for both sexes, it is unlikely that the international differences are due to occupational exposures, which usually vary according to gender, " said co-author Sharif B. Mohr, M.P.H.
Monday, September 18, 2006
More on Northern vs. Southern Europeans study
In my cursory reading of the paper from this previous post, I missed this, which I later found in a news story from the Texas A&M Anthropology in the News website:
They looked at SNPs associated with rheumatoid arthritis and found that, when they corrected for ancestry, several of the genes that were previously believed to be good candidates for being involved in the disease were no longer candidates at all. They also corrected for ancestry in a data set looking at lactose intolerance. "When we did not control for differences in population structure, we got a lot of false associations," Seldin explained.
They looked at SNPs associated with rheumatoid arthritis and found that, when they corrected for ancestry, several of the genes that were previously believed to be good candidates for being involved in the disease were no longer candidates at all. They also corrected for ancestry in a data set looking at lactose intolerance. "When we did not control for differences in population structure, we got a lot of false associations," Seldin explained.
Sunday, September 17, 2006
Genes underlying skin pigmentation differences
can't find the full text for this, ...not too sure what they do other than find recent positive selection on the DCT gene in Chinese... and that "it is likely that different genes are responsible for the lighter skin pigmentation found in different non-African populations." --- (with all respect, does this kind of sentence really need to be said at the end of an abstract. I mean- is it anything that isn't already obvious??)
more details on this once I get the full-text:
Identifying genes underlying skin pigmentation differences among human populations.
Myles S, Somel M, Tang K, Kelso J, Stoneking M
Hum Genet. 2006 Sep 15; [Epub ahead of print]
Abstract: Skin pigmentation is a human phenotype that varies greatly among human populations and it has long been speculated that this variation is adaptive. We therefore expect the genes that contribute to these large differences in phenotype to show large allele frequency differences among populations and to possibly harbor signatures of positive selection. To identify the loci that likely contribute to among-population human skin pigmentation differences, we measured allele frequency differentiation among Europeans, Chinese and Africans for 24 human pigmentation genes from 2 publicly available, large scale SNP data sets. Several skin pigmentation genes show unusually large allele frequency differences among these populations. To determine whether these allele frequency differences might be due to selection, we employed a within-population test based on long-range haplotype structure and identified several outliers that have not been previously identified as putatively adaptive. Most notably, we identify the DCT gene as a candidate for recent positive selection in the Chinese. Moreover, our analyses suggest that it is likely that different genes are responsible for the lighter skin pigmentation found in different non-African populations.
more details on this once I get the full-text:
Identifying genes underlying skin pigmentation differences among human populations.
Myles S, Somel M, Tang K, Kelso J, Stoneking M
Hum Genet. 2006 Sep 15; [Epub ahead of print]
Abstract: Skin pigmentation is a human phenotype that varies greatly among human populations and it has long been speculated that this variation is adaptive. We therefore expect the genes that contribute to these large differences in phenotype to show large allele frequency differences among populations and to possibly harbor signatures of positive selection. To identify the loci that likely contribute to among-population human skin pigmentation differences, we measured allele frequency differentiation among Europeans, Chinese and Africans for 24 human pigmentation genes from 2 publicly available, large scale SNP data sets. Several skin pigmentation genes show unusually large allele frequency differences among these populations. To determine whether these allele frequency differences might be due to selection, we employed a within-population test based on long-range haplotype structure and identified several outliers that have not been previously identified as putatively adaptive. Most notably, we identify the DCT gene as a candidate for recent positive selection in the Chinese. Moreover, our analyses suggest that it is likely that different genes are responsible for the lighter skin pigmentation found in different non-African populations.
Friday, September 15, 2006
Northern vs. Southern Europeans

Here, the authors type over 5000 SNPs in about 1000 Europeans and European Americans, use STRUCTURE, and find clustering that corresponds to Northern vs. Southern Europeans. Among other findings, the Finns clustered very strongly, Ashkenazi and Sephardic Jews clustered with the Southern Europeans.
Disease differences among Europeans according to authors: type 1 diabetes, Crohn' s disease, multiple sclerosis. (but also celiac disease, malaria, cystic fibrosis??)
European Population Substructure: Clustering of Northern and Southern Populations
Michael F. Seldin, Russell Shigeta, Pablo Villoslada, Carlo Selmi, Jaakko Tuomilehto, Gabriel Silva, John W. Belmont, Lars Klareskog, Peter K. Gregersen
PLoS Genetics: Volume 2 | Issue 9 | SEPTEMBER 2006
Synopsis: Two unrelated persons in the human population have hundreds of thousands of base pair differences between them in DNA sequence. Previous studies have shown that a small proportion of these sequence differences correlate with a person's continental ancestry: broadly, Asia, Africa Oceana, America, or continental Europe. In the current study, DNA differences within a particular continental group, Europe, were examined. Overall, the analysis of sequence variation allowed the authors to distinguish individuals with northern European ancestry (Swedish, English, Irish, German, and Ukrainian) from individuals with southern European ancestry (Italian, Spanish, Portuguese, and Greek). Interestingly, Ashkenazi Jewish individuals tend to group together with individuals from southern European countries. This study is important because it provides a method of taking into account these differences when searching for genetic variations that are associated with particular human traits, such as disease susceptibility, response to drug treatment, or side effects from therapy. Specifically, these methods may allow scientists to uncover disease-associated genetic variations that might be hidden unless differences related to European ancestry are considered.
Thursday, September 14, 2006
Genetic oddities of the vole
 from ScienceBlog.com, a story about a paper in Genetica by J.A. DeWoody about how voles have many genetic oddities. here's an excerpt:
from ScienceBlog.com, a story about a paper in Genetica by J.A. DeWoody about how voles have many genetic oddities. here's an excerpt:The study focuses on 60 species within the vole genus Microtus, which has evolved in the last 500,000 to 2 million years. This means voles are evolving 60-100 times faster than the average vertebrate in terms of creating different species. Within the genus (the level of taxonomic classification above species), the number of chromosomes in voles ranges from 17-64. DeWoody said that this is an unusual finding, since species within a single genus often have the same chromosome number.
Among the vole's other bizarre genetic traits:
•In one species, the X chromosome, one of the two sex-determining chromosomes (the other being the Y), contains about 20 percent of the entire genome. Sex chromosomes normally contain much less genetic information.
•In another species, females possess large portions of the Y (male) chromosome.
•In yet another species, males and females have different chromosome numbers, which is uncommon in animals.
A final "counterintuitive oddity" is that despite genetic variation, all voles look alike, said DeWoody's former graduate student and study co-author Deb Triant.
"All voles look very similar, and many species are completely indistinguishable," DeWoody said.
In one particular instance, DeWoody was unable to differentiate between two species even after close examination and analysis of their cranial structure; only genetic tests could reveal the difference.
Nevertheless, voles are perfectly adept at recognizing those of their own species.
"I have seen absolutely no evidence of mating between different species," Triant said. "We don't know how they do this, but scent and behavior probably play a role."
DeWoody said, "The vole is a great a model system that could be used to study lots of natural phenomena that could impact humans."
Tuesday, September 12, 2006
Common Errors in Ancient DNA sequencing
This is the paper that I referred to in a post last week about S. Paabo. Apparently, he is now a member of the National Academy of Sciences.
Patterns of nucleotide misincorporations during enzymatic amplification and direct large-scale sequencing of ancient DNA
M. Stiller, R. E. Green, M. Ronan, J. F. Simons, L. Du, W. He, M. Egholm, J. M. Rothberg, S. G. Keats, N. D. Ovodov, E. E. Antipina, G. F. Baryshnikov, Y. V. Kuzmin, A. A. Vasilevski, G. E. Wuenschell, J. Termini, M. Hofreiter, V. Jaenicke-Després, and S. Pääbo
PNAS 2006 103: 13578-13584
Abstract: Whereas evolutionary inferences derived from present-day DNA sequences are by necessity indirect, ancient DNA sequences provide a direct view of past genetic variants. However, base lesions that accumulate in DNA over time may cause nucleotide misincorporations when ancient DNA sequences are replicated. By repeated amplifications of mitochondrial DNA sequences from a large number of ancient wolf remains, we show that C/G-to-T/A transitions are the predominant type of such misincorporations. Using a massively parallel sequencing method that allows large numbers of single DNA strands to be sequenced, we show that modifications of C, as well as to a lesser extent of G, residues cause such misincorporations. Experiments where oligonucleotides containing modified bases are used as templates in amplification reactions suggest that both of these types of misincorporations can be caused by deamination of the template bases. New DNA sequencing methods in conjunction with knowledge of misincorporation processes have now, in principle, opened the way for the determination of complete genomes from organisms that became extinct during and after the last glaciation.
Patterns of nucleotide misincorporations during enzymatic amplification and direct large-scale sequencing of ancient DNA
M. Stiller, R. E. Green, M. Ronan, J. F. Simons, L. Du, W. He, M. Egholm, J. M. Rothberg, S. G. Keats, N. D. Ovodov, E. E. Antipina, G. F. Baryshnikov, Y. V. Kuzmin, A. A. Vasilevski, G. E. Wuenschell, J. Termini, M. Hofreiter, V. Jaenicke-Després, and S. Pääbo
PNAS 2006 103: 13578-13584
Abstract: Whereas evolutionary inferences derived from present-day DNA sequences are by necessity indirect, ancient DNA sequences provide a direct view of past genetic variants. However, base lesions that accumulate in DNA over time may cause nucleotide misincorporations when ancient DNA sequences are replicated. By repeated amplifications of mitochondrial DNA sequences from a large number of ancient wolf remains, we show that C/G-to-T/A transitions are the predominant type of such misincorporations. Using a massively parallel sequencing method that allows large numbers of single DNA strands to be sequenced, we show that modifications of C, as well as to a lesser extent of G, residues cause such misincorporations. Experiments where oligonucleotides containing modified bases are used as templates in amplification reactions suggest that both of these types of misincorporations can be caused by deamination of the template bases. New DNA sequencing methods in conjunction with knowledge of misincorporation processes have now, in principle, opened the way for the determination of complete genomes from organisms that became extinct during and after the last glaciation.
Sunday, September 10, 2006
A new panel of SNPs for admixture mapping in African Americans
A genomewide single-nucleotide-polymorphism panel with high ancestry information for african american admixture mapping.
Tian C, Hinds DA, Shigeta R, Kittles R, Ballinger DG, Seldin MF.
American Journal of Human Genetics. 2006 Oct;79(4):640-9. Epub 2006 Aug 15
Abstract: Admixture mapping requires a genomewide panel of relatively evenly spaced markers that can distinguish the ancestral origins of chromosomal segments in admixed individuals. Through use of the results of the International HapMap Project and specific selection criteria, the current study has examined the ability of selected single-nucleotide polymorphisms (SNPs) to extract continental ancestry information in African American subjects and to explore parameters for admixture mapping. Genotyping of two linguistically diverse West African populations (Bini and Kanuri Nigerians, who are Niger-Congo [Bantu] and Nilo-Saharan speakers, respectively), European Americans, and African Americans validated a genomewide set of >4,000 SNP ancestry-informative markers with mean and median F(ST) values >0.59 and mean and median Fisher's information content >2.5. This set of SNPs extracted a larger amount of ancestry information in African Americans than previously reported SNP panels and provides nearly uniform coverage of the genome. Moreover, in the current study, simulations show that this more informative panel improves power for admixture mapping in African Americans when ethnicity risk ratios are modest. This is particularly important in the application of admixture mapping in complex genetic diseases for which only modest ethnicity risk ratios of relevant susceptibility genes are expected.
Tian C, Hinds DA, Shigeta R, Kittles R, Ballinger DG, Seldin MF.
American Journal of Human Genetics. 2006 Oct;79(4):640-9. Epub 2006 Aug 15
Abstract: Admixture mapping requires a genomewide panel of relatively evenly spaced markers that can distinguish the ancestral origins of chromosomal segments in admixed individuals. Through use of the results of the International HapMap Project and specific selection criteria, the current study has examined the ability of selected single-nucleotide polymorphisms (SNPs) to extract continental ancestry information in African American subjects and to explore parameters for admixture mapping. Genotyping of two linguistically diverse West African populations (Bini and Kanuri Nigerians, who are Niger-Congo [Bantu] and Nilo-Saharan speakers, respectively), European Americans, and African Americans validated a genomewide set of >4,000 SNP ancestry-informative markers with mean and median F(ST) values >0.59 and mean and median Fisher's information content >2.5. This set of SNPs extracted a larger amount of ancestry information in African Americans than previously reported SNP panels and provides nearly uniform coverage of the genome. Moreover, in the current study, simulations show that this more informative panel improves power for admixture mapping in African Americans when ethnicity risk ratios are modest. This is particularly important in the application of admixture mapping in complex genetic diseases for which only modest ethnicity risk ratios of relevant susceptibility genes are expected.
Friday, September 08, 2006
MC1R and melanoma risk: what's the pathway?
(look at all the greek author names!!)
This paper examines the association between melanoma and MC1R variants in 123 melanoma patients and 155 control subjects from Greece. In general, MC1R variants were associated with higher melanoma risk, yet it's still not known what kind of pathway is operating here (through pigment related pathway, or another pathway that MC1R may be involved in).
There is also a commentary by Jonathan Rees that accompanies this paper, with this interesting passage:
Even though we may be uncertain as to the nature of the pathways linking MC1Rwith cancer risk, surprising though it may seem, it is still possible to assess the relative quantitative contributions of such alternative pathways. In a critical study, Dwyer and colleagues studied a group of patients with melanoma and control subjects, and, besides carrying out MC1R sequencing, they also assayed the pigmentary phenotype by measuring skin reflectance on the upper inner arm (Dwyer et al., 2004). They then used a now fairly standard receiver operating characteristic (ROC) curve analysis to estimate how much information sequencing provided over and above that provided by skin color. They found that the additional information provided by MC1R was greater for basal-cell carcinoma than for melanoma, but even for basal-cell carcinoma the amount of information provided was modest. So, if age and sex could predict 42 of the 640 cases of basal-cell carcinoma expected in a cohort of 10,000 over 10 years, skin reflectance allowed prediction of a further 14 cases (56 in total), and adding in MC1R sequence would only add another two cases (58 in total). Why is this important to our interpretation of the data presented by Stratigos et al. (2006)? First, it suggests that any putative pathways linking MC1R variation and cancer risk (beyond skin color) are numerically small. Second, and of much greater importance, it serves to remind us that although we may identify and report modest odds ratios between genes and a disease state, in many instances we are perhaps an order of magnitude out in our ability to provide predictions of a future disease state that are meaningful for individual patients. So whereas our accumulating knowledge might gradually allow screening for melanoma to be considered, one has the feeling that there is a whole chunk of pigmentary biology that is still invisible to us. It seems, therefore, that some chapters of this particular story remain to be written.
Melanocortin Receptor-1 Gene Polymorphisms and the Risk of Cutaneous Melanoma in a Low-Risk Southern European Population
Alexander J Stratigos, Gerasimos Dimisianos, Vasiliki Nikolaou, Mirto Poulou, Vana Sypsa, Irene Stefanaki, Othon Papadopoulos, Dorothea Polydorou, Michaela Plaka, Eleftheria Christofidou, Helen Gogas, Dimosthenis Tsoutsos, Ourania Kastana, Christina Antoniou, Angelos Hatzakis, Emmanouil Kanavakis and Andreas D Katsambas
Journal of Investigative Dermatology (2006) 126, 1842–1849. doi:10.1038/sj.jid.5700292; published online 6 April 2006
Abstract: Individuals with melanocortin 1 receptor (MC1R) gene variants have been shown to carry an increased risk for the development of melanoma. In this study, we investigated the relationship of MC1R gene variants and the risk of melanoma in 123 melanoma patients and 155 control subjects from Greece. The entire MC1R gene was sequenced for polymorphisms and the results were correlated with host factors and pigmentary characteristics. MC1R polymorphisms were present in 59.4% of melanoma patients compared to 37.5% of controls, yielding an odds ratio (OR) of 2.43 (95% confidence interval (CI)=1.50–3.96, P<0.001) or="6.97;" ci="1.86–26.12,">P=0.004). Only the Val60Leu, Arg142His, and Arg151Cys variants were significantly associated with melanoma risk. In stratified analysis, the risk of melanoma among MC1R carriers was not influenced by skin phototype, skin color, or hair color. No association was found between MC1R genotype and the age of onset of melanoma, the tumor location, or the tumor thickness. In conclusion, MC1R polymorphisms are a predisposing factor of melanoma in a southern European population with a relatively low incidence of the disease.
This paper examines the association between melanoma and MC1R variants in 123 melanoma patients and 155 control subjects from Greece. In general, MC1R variants were associated with higher melanoma risk, yet it's still not known what kind of pathway is operating here (through pigment related pathway, or another pathway that MC1R may be involved in).
There is also a commentary by Jonathan Rees that accompanies this paper, with this interesting passage:
Even though we may be uncertain as to the nature of the pathways linking MC1Rwith cancer risk, surprising though it may seem, it is still possible to assess the relative quantitative contributions of such alternative pathways. In a critical study, Dwyer and colleagues studied a group of patients with melanoma and control subjects, and, besides carrying out MC1R sequencing, they also assayed the pigmentary phenotype by measuring skin reflectance on the upper inner arm (Dwyer et al., 2004). They then used a now fairly standard receiver operating characteristic (ROC) curve analysis to estimate how much information sequencing provided over and above that provided by skin color. They found that the additional information provided by MC1R was greater for basal-cell carcinoma than for melanoma, but even for basal-cell carcinoma the amount of information provided was modest. So, if age and sex could predict 42 of the 640 cases of basal-cell carcinoma expected in a cohort of 10,000 over 10 years, skin reflectance allowed prediction of a further 14 cases (56 in total), and adding in MC1R sequence would only add another two cases (58 in total). Why is this important to our interpretation of the data presented by Stratigos et al. (2006)? First, it suggests that any putative pathways linking MC1R variation and cancer risk (beyond skin color) are numerically small. Second, and of much greater importance, it serves to remind us that although we may identify and report modest odds ratios between genes and a disease state, in many instances we are perhaps an order of magnitude out in our ability to provide predictions of a future disease state that are meaningful for individual patients. So whereas our accumulating knowledge might gradually allow screening for melanoma to be considered, one has the feeling that there is a whole chunk of pigmentary biology that is still invisible to us. It seems, therefore, that some chapters of this particular story remain to be written.
Melanocortin Receptor-1 Gene Polymorphisms and the Risk of Cutaneous Melanoma in a Low-Risk Southern European Population
Alexander J Stratigos, Gerasimos Dimisianos, Vasiliki Nikolaou, Mirto Poulou, Vana Sypsa, Irene Stefanaki, Othon Papadopoulos, Dorothea Polydorou, Michaela Plaka, Eleftheria Christofidou, Helen Gogas, Dimosthenis Tsoutsos, Ourania Kastana, Christina Antoniou, Angelos Hatzakis, Emmanouil Kanavakis and Andreas D Katsambas
Journal of Investigative Dermatology (2006) 126, 1842–1849. doi:10.1038/sj.jid.5700292; published online 6 April 2006
Abstract: Individuals with melanocortin 1 receptor (MC1R) gene variants have been shown to carry an increased risk for the development of melanoma. In this study, we investigated the relationship of MC1R gene variants and the risk of melanoma in 123 melanoma patients and 155 control subjects from Greece. The entire MC1R gene was sequenced for polymorphisms and the results were correlated with host factors and pigmentary characteristics. MC1R polymorphisms were present in 59.4% of melanoma patients compared to 37.5% of controls, yielding an odds ratio (OR) of 2.43 (95% confidence interval (CI)=1.50–3.96, P<0.001) or="6.97;" ci="1.86–26.12,">P=0.004). Only the Val60Leu, Arg142His, and Arg151Cys variants were significantly associated with melanoma risk. In stratified analysis, the risk of melanoma among MC1R carriers was not influenced by skin phototype, skin color, or hair color. No association was found between MC1R genotype and the age of onset of melanoma, the tumor location, or the tumor thickness. In conclusion, MC1R polymorphisms are a predisposing factor of melanoma in a southern European population with a relatively low incidence of the disease.
Thursday, September 07, 2006
MALD for prostate cancer in African Americans
First a short description of MALD from the paper cited below:
The idea of admixture mapping is to screen through the genome of populations of mixed ancestry such as African American, searching for regions where the proportion of DNA inherited from either the ancestral European or African population is unusual compared with the genome wide average. Admixture mapping requires a relatively small number of markers for a whole genome scan: a couple of thousand, rather than the hundreds of thousands estimated to be necessary in nonadmixed populations. Because the mixture between European and West-African populations occured within the past 15 generations, stretches of DNA with contiguous European and African ancestry have not had much time to break up because of recombination and typically extend millions of base pairs. Admixture mapping therefore studies highly selected SNPs every few million base pairs, rather than every few thousand as with linkage disequilibrium mapping.
and one of the main conclusions from this study:
...we show that the 8q24 locus contributes to a major increased risk for prostate cancer in African Americans with African ancestry at 8q24. The difference between these individuals and African Americans with European ancestry at 8q24 explains a large proportion of prostate cancer in younger African Americans.
Admixture mapping identifies 8q24 as a prostate cancer risk locus in African American men
Matthew L. Freedman,Christopher A. Haiman, Nick Patterson, Gavin J. McDonald, Arti Tandon, Alicja Waliszewska, Kathryn Penney, Robert G. Steen, Kristin Ardlie, Esther M. John, Ingrid Oakley-Girvan, Alice S. Whittemore, Kathleen A. Cooney, Sue A. Ingles, David Altshuler, Brian E. Henderson, and David Reich
PNAS: Early Edition
Abstract: A whole-genome admixture scan in 1,597 >African Americans identified a 3.8 Mb interval on chromosome 8q24 as significantly associated with susceptibility to prostate cancer [logarithm of odds (LOD) = 7.1]. The increased risk because of inheriting African ancestry is greater in men diagnosed before 72 years of age (P <> and may contribute to the epidemiological observation that the higher risk for prostate cancer in African Americans is greatest in younger men (and attenuates with older age). The same region was recently identified through linkage analysis of prostate cancer, followed by fine-mapping. We strongly replicated this association (P <>-9) but find that the previously described alleles do not explain more than a fraction of the admixture signal. Thus, admixture mapping indicates a major, still-unidentified risk gene for prostate cancer at 8q24, motivating intense work to find it.
The idea of admixture mapping is to screen through the genome of populations of mixed ancestry such as African American, searching for regions where the proportion of DNA inherited from either the ancestral European or African population is unusual compared with the genome wide average. Admixture mapping requires a relatively small number of markers for a whole genome scan: a couple of thousand, rather than the hundreds of thousands estimated to be necessary in nonadmixed populations. Because the mixture between European and West-African populations occured within the past 15 generations, stretches of DNA with contiguous European and African ancestry have not had much time to break up because of recombination and typically extend millions of base pairs. Admixture mapping therefore studies highly selected SNPs every few million base pairs, rather than every few thousand as with linkage disequilibrium mapping.
and one of the main conclusions from this study:
...we show that the 8q24 locus contributes to a major increased risk for prostate cancer in African Americans with African ancestry at 8q24. The difference between these individuals and African Americans with European ancestry at 8q24 explains a large proportion of prostate cancer in younger African Americans.
Admixture mapping identifies 8q24 as a prostate cancer risk locus in African American men
Matthew L. Freedman,
PNAS: Early Edition
Abstract:
Wednesday, September 06, 2006
A SNP implicated in preterm birth among African Americans
I thought this was a great paper because it investigated the phenomena from several angles:
- differences in genotype between many ancestry groups (including many Sierra Leone groups, for some reason)
- tissue culture and transfection
- EMSA
- population structure of study group
... and more
A functional SNP in the promoter of the SERPINH1 gene increases risk of premature rupture of membranes in African Americans
Hongyan Wang, Samuel Parry, George Macones, Mary D. Sammel, Helena Kuivaniemi, Gerard Tromp, George Argyropoulos, Indrani Halder, Mark D. Shriver, Roberto Romero, and Jerome F. Strauss, III
Abstract: Prematurity is more prevalent in African Americans than in European Americans. We investigated the contribution of a functional SNP in the promoter of the SERPINH1 gene, enriched among those of African ancestry, to preterm premature rupture of membranes (PPROM), the leading identifiable cause of preterm birth. SERPINH1 encodes heat-shock protein 47, a chaperone essential for collagen synthesis. The SERPINH1 –656 minor T allele had a greater frequency in African populations and African Americans than in European Americans (12.4% vs. 4.1%). The –656 T allele displayed significantly reduced promoter activity compared to the major –656 C allele in amnion fibroblasts, which lay down the fibrillar collagen that gives tensile strength to the amnion. An initial case-control study demonstrated that the –656 T allele is significantly more frequent in African-American neonates (P <> PPROM compared with controls (odds ratio of 3.22, 95% confidence interval 1.50, 7.22). There was no significant difference in ancestry among cases and controls using a dihybrid model based on 29 ancestry-informative markers. Adjusting the results of the case-control study for admixture still yielded a statistically significant association between the –656 T allele and PPROM (P <> results. The combined case-control findings showed a highly significant (P <> T allele and PPROM. The SERPINH1 –656 T allele is the first example of an ancestry-informative marker associated with preterm birth in African Americans.
- differences in genotype between many ancestry groups (including many Sierra Leone groups, for some reason)
- tissue culture and transfection
- EMSA
- population structure of study group
... and more
A functional SNP in the promoter of the SERPINH1 gene increases risk of premature rupture of membranes in African Americans
Hongyan Wang, Samuel Parry, George Macones, Mary D. Sammel, Helena Kuivaniemi, Gerard Tromp, George Argyropoulos, Indrani Halder, Mark D. Shriver, Roberto Romero, and Jerome F. Strauss, III
PNAS Sept. 5, 2006; 103:13463-13467
Abstract: Prematurity is more prevalent in African Americans than in European Americans. We investigated the contribution of a functional SNP in the promoter of the SERPINH1 gene, enriched among those of African ancestry, to preterm premature rupture of membranes (PPROM), the leading identifiable cause of preterm birth. SERPINH1 encodes heat-shock protein 47, a chaperone essential for collagen synthesis. The SERPINH1 –656 minor T allele had a greater frequency in African populations and African Americans than in European Americans (12.4% vs. 4.1%). The –656 T allele displayed significantly reduced promoter activity compared to the major –656 C allele in amnion fibroblasts, which lay down the fibrillar collagen that gives tensile strength to the amnion. An initial case-control study demonstrated that the –656 T allele is significantly more frequent in African-American neonates (P <> PPROM compared with controls (odds ratio of 3.22, 95% confidence interval 1.50, 7.22). There was no significant difference in ancestry among cases and controls using a dihybrid model based on 29 ancestry-informative markers. Adjusting the results of the case-control study for admixture still yielded a statistically significant association between the –656 T allele and PPROM (P <> results. The combined case-control findings showed a highly significant (P <> T allele and PPROM. The SERPINH1 –656 T allele is the first example of an ancestry-informative marker associated with preterm birth in African Americans.
Tuesday, September 05, 2006
Svante Paabo profile
PNAS has a profile of Svante Paabo in the early edition. Apparently he has a paper coming out soon in PNAS (his "Inaugural Article") about how damage to DNA can cause sequencing errors.
The profile is quite interesting ...traces his career and talks briefly about some of his major findings and areas of future interest.
The most exciting thing now is the Neanderthal autosomal, and ancient DNA sequencing...but also chimp human differences ("gene expression vs. gene sequence"): positive selection in the male germ line (never heard of this, not sure exactly what it means) and in brain related genes.
here is the PDF for those subscribed.
The profile is quite interesting ...traces his career and talks briefly about some of his major findings and areas of future interest.
The most exciting thing now is the Neanderthal autosomal, and ancient DNA sequencing...but also chimp human differences ("gene expression vs. gene sequence"): positive selection in the male germ line (never heard of this, not sure exactly what it means) and in brain related genes.
here is the PDF for those subscribed.
Sunday, September 03, 2006
More on structural variation in the genome
Unfortunately, I don't have access to the full text, but it looks like this paper is another good review of "various structural rearrangements .... with their influence on human phenotypic variation."
Structural Variation of the Human Genome
Andrew J. Sharp, Ze Cheng, and Evan E. Eichler
There is growing appreciation that the human genome contains significant numbers of structural rearrangements, such as insertions, deletions, inversions, and large tandem repeats. Recent studies have defined approximately 5% of the human genome as structurally variant in the normal population, involving more than 800 independent genes. We present a detailed review of the various structural rearrangements identified to date in humans, with particular reference to their influence on human phenotypic variation. Our current knowledge of the extent of human structural variation shows that the human genome is a highly dynamic structure that shows significant large-scale variation from the currently published genome reference sequence.
Structural Variation of the Human Genome
Subscribe to:
Comments (Atom)

