A worldwide survey of haplotype variation and linkage disequilibrium in the human genome
Donald F Conrad, Mattias Jakobsson, Graham Coop, Xiaoquan Wen, Jeffrey D Wall, Noah A Rosenberg & Jonathan K Pritchard
Nature Genetics 38, 1251 - 1260 (2006)
Abstract: Recent genomic surveys have produced high-resolution haplotype information, but only in a small number of human populations. We report haplotype structure across 12 Mb of DNA sequence in 927 individuals representing 52 populations. The geographic distribution of haplotypes reflects human history, with a loss of haplotype diversity as distance increases from Africa. Although the extent of linkage disequilibrium (LD) varies markedly across populations, considerable sharing of haplotype structure exists, and inferred recombination hotspot locations generally match across groups. The four samples in the International HapMap Project contain the majority of common haplotypes found in most populations: averaging across populations, 83% of common 20-kb haplotypes in a population are also common in the most similar HapMap sample. Consequently, although the portability of tag SNPs based on the HapMap is reduced in low-LD Africans, the HapMap will be helpful for the design of genome-wide association mapping studies in nearly all human populations.
Tuesday, October 31, 2006
Monday, October 30, 2006
Martinez-Marignac VL, Valladares A, Cameron E, Chan A, Perera A, Globus-Goldberg R, Wacher N, Kumate J, McKeigue P, O'donnell D, Shriver MD, Cruz M, Parra EJ.
Hum Genet. 2006 Oct 26; [Epub ahead of print]
Abstract: Admixture mapping is a recently developed method for identifying genetic risk factors involved in complex traits or diseases showing prevalence differences between major continental groups. Type 2 diabetes (T2D) is at least twice as prevalent in Native American populations as in populations of European ancestry, so admixture mapping is well suited to study the genetic basis of this complex disease. We have characterized the admixture proportions in a sample of 286 unrelated T2D patients and 275 controls from Mexico City and we discuss the implications of the results for admixture mapping studies. Admixture proportions were estimated using 69 autosomal ancestry-informative markers (AIMs). Maternal and paternal contributions were estimated from geographically informative mtDNA and Y-specific polymorphisms. The average proportions of Native American, European and, West African admixture were estimated as 65, 30, and 5%, respectively. The contributions of Native American ancestors to maternal and paternal lineages were estimated as 90 and 40%, respectively. In a logistic model with higher educational status as dependent variable, the odds ratio for higher educational status associated with an increase from 0 to 1 in European admixture proportions was 9.4 (95%, credible interval 3.8-22.6). This association of socioeconomic status with individual admixture proportion shows that genetic stratification in this population is paralleled, and possibly maintained, by socioeconomic stratification. The effective number of generations back to unadmixed ancestors was 6.7 (95% CI 5.7-8.0), from which we can estimate that genome-wide admixture mapping will require typing about 1,400 evenly distributed AIMs to localize genes underlying disease risk between populations of European and Native American ancestry. Sample sizes of about 2,000 cases will be required to detect any locus that contributes an ancestry risk ratio of at least 1.5.
Hum Genet. 2006 Oct 26; [Epub ahead of print]
Abstract: Admixture mapping is a recently developed method for identifying genetic risk factors involved in complex traits or diseases showing prevalence differences between major continental groups. Type 2 diabetes (T2D) is at least twice as prevalent in Native American populations as in populations of European ancestry, so admixture mapping is well suited to study the genetic basis of this complex disease. We have characterized the admixture proportions in a sample of 286 unrelated T2D patients and 275 controls from Mexico City and we discuss the implications of the results for admixture mapping studies. Admixture proportions were estimated using 69 autosomal ancestry-informative markers (AIMs). Maternal and paternal contributions were estimated from geographically informative mtDNA and Y-specific polymorphisms. The average proportions of Native American, European and, West African admixture were estimated as 65, 30, and 5%, respectively. The contributions of Native American ancestors to maternal and paternal lineages were estimated as 90 and 40%, respectively. In a logistic model with higher educational status as dependent variable, the odds ratio for higher educational status associated with an increase from 0 to 1 in European admixture proportions was 9.4 (95%, credible interval 3.8-22.6). This association of socioeconomic status with individual admixture proportion shows that genetic stratification in this population is paralleled, and possibly maintained, by socioeconomic stratification. The effective number of generations back to unadmixed ancestors was 6.7 (95% CI 5.7-8.0), from which we can estimate that genome-wide admixture mapping will require typing about 1,400 evenly distributed AIMs to localize genes underlying disease risk between populations of European and Native American ancestry. Sample sizes of about 2,000 cases will be required to detect any locus that contributes an ancestry risk ratio of at least 1.5.
Saturday, October 28, 2006
Smithsonian makes a statement
Check out Anthropology.net for a story on how the Smithsonian is refusing to display the Lucy skeleton that will be making a US tour over the next several years. Basically, the risk involved in moving this skeleton is deemed too high.
Friday, October 27, 2006
Cooperation model allows for adjustment of social ties
Cooperation Prevails When Individuals Adjust Their Social Ties
Francisco C. Santos, Jorge M. Pacheco, Tom Lenaerts
PLoS Computuational Biology Volume 2 | Issue 10 | OCTOBER 2006
Abstract: Conventional evolutionary game theory predicts that natural selection favours the selfish and strong even though cooperative interactions thrive at all levels of organization in living systems. Recent investigations demonstrated that a limiting factor for the evolution of cooperative interactions is the way in which they are organized, cooperators becoming evolutionarily competitive whenever individuals are constrained to interact with few others along the edges of networks with low average connectivity. Despite this insight, the conundrum of cooperation remains since recent empirical data shows that real networks exhibit typically high average connectivity and associated single-to-broad–scale heterogeneity. Here, a computational model is constructed in which individuals are able to self-organize both their strategy and their social ties throughout evolution, based exclusively on their self-interest. We show that the entangled evolution of individual strategy and network structure constitutes a key mechanism for the sustainability of cooperation in social networks. For a given average connectivity of the population, there is a critical value for the ratio W between the time scales associated with the evolution of strategy and of structure above which cooperators wipe out defectors. Moreover, the emerging social networks exhibit an overall heterogeneity that accounts very well for the diversity of patterns recently found in acquired data on social networks. Finally, heterogeneity is found to become maximal when W reaches its critical value. These results show that simple topological dynamics reflecting the individual capacity for self-organization of social ties can produce realistic networks of high average connectivity with associated single-to-broad–scale heterogeneity. On the other hand, they show that cooperation cannot evolve as a result of “social viscosity” alone in heterogeneous networks with high average connectivity, requiring the additional mechanism of topological co-evolution to ensure the survival of cooperative behaviour.
Francisco C. Santos, Jorge M. Pacheco, Tom Lenaerts
PLoS Computuational Biology Volume 2 | Issue 10 | OCTOBER 2006
Abstract: Conventional evolutionary game theory predicts that natural selection favours the selfish and strong even though cooperative interactions thrive at all levels of organization in living systems. Recent investigations demonstrated that a limiting factor for the evolution of cooperative interactions is the way in which they are organized, cooperators becoming evolutionarily competitive whenever individuals are constrained to interact with few others along the edges of networks with low average connectivity. Despite this insight, the conundrum of cooperation remains since recent empirical data shows that real networks exhibit typically high average connectivity and associated single-to-broad–scale heterogeneity. Here, a computational model is constructed in which individuals are able to self-organize both their strategy and their social ties throughout evolution, based exclusively on their self-interest. We show that the entangled evolution of individual strategy and network structure constitutes a key mechanism for the sustainability of cooperation in social networks. For a given average connectivity of the population, there is a critical value for the ratio W between the time scales associated with the evolution of strategy and of structure above which cooperators wipe out defectors. Moreover, the emerging social networks exhibit an overall heterogeneity that accounts very well for the diversity of patterns recently found in acquired data on social networks. Finally, heterogeneity is found to become maximal when W reaches its critical value. These results show that simple topological dynamics reflecting the individual capacity for self-organization of social ties can produce realistic networks of high average connectivity with associated single-to-broad–scale heterogeneity. On the other hand, they show that cooperation cannot evolve as a result of “social viscosity” alone in heterogeneous networks with high average connectivity, requiring the additional mechanism of topological co-evolution to ensure the survival of cooperative behaviour.
Thursday, October 26, 2006
Brain size and diet in orangutans
 This is a paper in press in the Journal of Human Evolution. They found that individuals in one well fed group of orangs had larger brain sizes than in another not so well fed group. They say that this lends some support to the expensive tissue hypothesis. The sample sizes are small and, if I'm not mistaken, the differences are only significant for females. From the abstract (the link to H. floresiensis is interesting):
This is a paper in press in the Journal of Human Evolution. They found that individuals in one well fed group of orangs had larger brain sizes than in another not so well fed group. They say that this lends some support to the expensive tissue hypothesis. The sample sizes are small and, if I'm not mistaken, the differences are only significant for females. From the abstract (the link to H. floresiensis is interesting):Our results, therefore, provide conditional support for the hypothesis that decreased brain size is related to prolonged episodes of food scarcity, and suggest a correlation between brain size, diet quality, and life history at the lowest macroevolutionary level. The association of a relatively small brain and poor diet quality in Pongo further suggests that ecological factors may plausibly account for such a reduction in brain size as observed in the recently recovered Homo floresiensis from Indonesia.
here's a press release from EurekAlert
Variation in brain size and ecology in Pongo
Andrea B. Taylor, and Carel P. van Schaik
Journal of Human Evolution Arcticle in Press
Abstract: Numerous hypotheses have been advanced to explain relative increases in brain size in primates and other mammals. However, notably less attention has been directed towards addressing the biological limits to increasing brain size. Here we explore variation in brain size in orangutans. We evaluated both raw and size-adjusted cranial capacity (CC) in adult Pongo pygmaeus pygmaeus (n = 147), P. p. wurmbii (n = 24), P. p. morio (n = 14), and P. abelii (n = 36). Results demonstrate significant variation in CC among orangutan taxa. Population differences in raw CC are significant for females (p = 0.014) but not males. Post-hoc pairwise comparisons among females further reveal that raw CC is significantly smaller in P. p. morio compared to both P. abelii and P. p. pygmaeus. When evaluated for proportionality, geometric equivalence in CC is not maintained in orangutans, as P. p. morio has a significantly smaller CC when compared to one or more other orangutan groups. Even after statistically partitioning size and size-correlated shape, P. p. morio has a significantly smaller CC compared to most other orangutan groups. These observed differences in relative brain size are consistent with known variation in resource quality and life history amongst orangutan populations. Specifically, P. p. morio is characterized by the least productive habitat, the lowest energy intake during extended lean periods, and the shortest interbirth intervals. Our results, therefore, provide conditional support for the hypothesis that decreased brain size is related to prolonged episodes of food scarcity, and suggest a correlation between brain size, diet quality, and life history at the lowest macroevolutionary level. The association of a relatively small brain and poor diet quality in Pongo further suggests that ecological factors may plausibly account for such a reduction in brain size as observed in the recently recovered Homo floresiensis from Indonesia.
Four Stone Hearth- Anthropology Blog Carnival
Check out the first edition of the Four Stone Hearth ... the first of a biweekly/bimonthly (whichever one means twice a month) Anthropology Blog Carnival...a sort of coming together of various Anthro-related blogs.
Tuesday, October 24, 2006
The uniqueness of Andaman Islanders
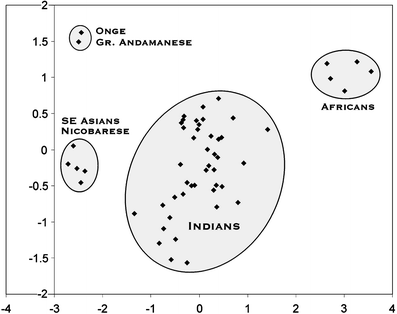 The authors use 9 autosomal STR markers to examine the affinities between The Andaman Islanders and Indians and Africans. I am unsure as to why they didn't examine other neighboring populations (Thailand, Indonesia, Burma), but maybe because in their previous study on Y-chromosome and mt-DNA they find the closest affinity with Indians. Anyway, they find/confirm that the Andaman Islanders are genetically very unique and may trace their ancestry to a first wave of humans out of Africa.
The authors use 9 autosomal STR markers to examine the affinities between The Andaman Islanders and Indians and Africans. I am unsure as to why they didn't examine other neighboring populations (Thailand, Indonesia, Burma), but maybe because in their previous study on Y-chromosome and mt-DNA they find the closest affinity with Indians. Anyway, they find/confirm that the Andaman Islanders are genetically very unique and may trace their ancestry to a first wave of humans out of Africa.Unique origin of Andaman Islanders: insight from autosomal loci
K. Thangaraj, G. Chaubey, A. G. Reddy, V. K. Sing and L. Singh
Journal of Human Genetics Volume 51, Number 9 / September, 2006
Abstract: Our mtDNA and Y chromosome studies lead to the conclusion that the Andamanese “Negrito” mtDNA lineages have survived in the Andaman Islands in complete genetic isolation from other South and Southeast Asian populations since the initial settlement of the region by the out-of-Africa migration. In order to obtain a robust reconstruction of the evolutionary history of the Andamanese, we carried out a study on the three aboriginal populations, namely, the Great Andamanese, Onge and Nicobarese, using autosomal microsatellite markers. The range of alleles (7–31.2) observed in the studied population and heterozygosity values (0.392–0.857) indicate that the selected STR markers are highly polymorphic in all the three populations, and genetic variability within the populations is significantly high, with a mean gene diversity of 77%. The Andaman “Negrito” populations do not show particular affinities either with the African populations or with the Indian populations, confirming their unique origin. In contrast, Nicobarese show close affinities with the Southeast Asian populations, suggesting their recent entry in the Islands.
Monday, October 23, 2006
ACTN3 and muscle function
 Variants of the ACTN3 gene show pretty large population differences (also not present in chimps) and have been associated with ability in sprint sports vs. endurance. This is thought to be due to the function of ACTN3 in determining the slow twitch vs. fast twitch muscle fiber proportions. In this paper they look at some Greek people (not a self-selected group, because unlike previous studies, these are not professional or even amateur athletes and they are young) to see if they can reproduce that association and they only find an association for the 40 meter sprint in the predicted direction. They cite a paper that I had also missed that fails to support the hypothesized association among Kenyan marathon runners.
Variants of the ACTN3 gene show pretty large population differences (also not present in chimps) and have been associated with ability in sprint sports vs. endurance. This is thought to be due to the function of ACTN3 in determining the slow twitch vs. fast twitch muscle fiber proportions. In this paper they look at some Greek people (not a self-selected group, because unlike previous studies, these are not professional or even amateur athletes and they are young) to see if they can reproduce that association and they only find an association for the 40 meter sprint in the predicted direction. They cite a paper that I had also missed that fails to support the hypothesized association among Kenyan marathon runners.Association analysis of the ACTN3 R577X polymorphism and complex quantitative body composition and performance phenotypes in adolescent Greeks
European Journal of Human Genetics
advance online publication 11 October 2006
Colin N Mora, Nan Yang, Mark E S Bailey, Athanasios Tsiokanos, Athanasios Jamurtas, Daniel G MacArthur, Kathryn North, Yannis P Pitsiladis and Richard H Wilson
Abstract: The functional allele (577R) of ACTN3, which encodes human -actinin-3, has been reported to be associated with elite athletic status and with response to resistance training, while the nonfunctional allele (577X) has been proposed as a candidate metabolically thrifty allele. In a study of 992 adolescent Greeks, we show that there is a significant association (P=0.003) between the ACTN3 R577X polymorphism and 40 m sprint time in males that accounts for 2.3% of phenotypic variance, with the 577R allele contributing to faster times in an additive manner. The R577X polymorphism is not associated with other power phenotypes related to 40 m sprint, nor with an endurance phenotype. Furthermore, the polymorphism is not associated with obesity-related phenotypes in our population, suggesting that the 577X allele is not a thrifty allele, and thus the persistence of this null allele must be explained in other terms.
Genetic relatedness in chimp groups
Check out John Hawks' entry on Patrilocality and genetic relatedness in chimpanzees. They fail to support the hypothesis that interactions in large groups are driven strongly by kin selection, because the relatedness between males of the same group is only a bit higher than relatedness between groups.
In the paper, the authors assume that humans are universally patrilocal, but this still seems to be under considerable debate. (once I'm organized, I'll start giving references for this sort of thing)
In the paper, the authors assume that humans are universally patrilocal, but this still seems to be under considerable debate. (once I'm organized, I'll start giving references for this sort of thing)
Friday, October 20, 2006
What it takes to become a good hunter
 To be fair, I haven't read the paper, but it seems like body size and skills/knowledge are not the only hypotheses that predict hunting competence. Another might just be motivation, mainly mediated by number of dependents. In addition, Kaplan et al. have found that hunting return rates tend to peak at 4o years old or so, long after human males have reached their peak body size, so I'm unclear as to why they would even consider something like body size.
To be fair, I haven't read the paper, but it seems like body size and skills/knowledge are not the only hypotheses that predict hunting competence. Another might just be motivation, mainly mediated by number of dependents. In addition, Kaplan et al. have found that hunting return rates tend to peak at 4o years old or so, long after human males have reached their peak body size, so I'm unclear as to why they would even consider something like body size.How long does it take to become a proficient hunter? Implications for the evolution of extended development and long life span
Michael Gurven, Hillard Kaplan and Maguin Gutierrez
Journal of Human Evolution Volume 51, Issue 5 , November 2006, Pages 454-470
Abstract: Human hunting is arguably one of the most difficult activities common to foraging peoples now and in the past. Children and teenagers have usually been described as incompetent hunters in ethnographies of hunter-gatherers. This paper explores the extent to which adult-level competence is limited more by the constraints of physical capital, or body size, and brain-based capital, or skills and learning. The grandmother hypothesis requires that production is an increasing function of size alone, while the embodied capital model stipulates that production is a function of both size and delayed learning. Tests based on observational, interview, and experimental data collected among Tsimane Amerindians of the Bolivian Amazon suggest that size alone cannot explain the long delay until peak hunting productivity. Indirect encounters (e.g., smells, sounds, tracks, and scat) and shooting of stationary targets are two components of hunting ability limited primarily by physical size alone, but the more difficult components of hunting—direct encounters with important prey items and successful capture—require substantial skill. Those skills can take an additional ten to twenty years to develop after achieving adult body size.
Wednesday, October 18, 2006
Characteristics of HARs (human accelerated regions)
...they are:
- A/T to G/C changes (weak to strong)
- located in high recombination areas
- next to genes that are enriched for transcription factors
- located in high G/C content areas near telomeres
Forces Shaping the Fastest Evolving Regions in the Human Genome
Katherine S. Pollard, Sofie R. Salama, Bryan King, Andrew D. Kern, Tim Dreszer, Sol Katzman1, Adam Siepel, Jakob S. Pedersen, Gill Bejerano, Robert Baertsch, Kate R. Rosenbloom, Jim Kent, David Haussler
PLoS Genetics vol. 2 issue 10; October 2006
Abstract: Comparative genomics allow us to search the human genome for segments that were extensively changed in the last ~5 million years since divergence from our common ancestor with chimpanzee, but are highly conserved in other species and thus are likely to be functional. We found 202 genomic elements that are highly conserved in vertebrates but show evidence of significantly accelerated substitution rates in human. These are mostly in non-coding DNA, often near genes associated with transcription and DNA binding. Resequencing confirmed that the five most accelerated elements are dramatically changed in human but not in other primates, with seven times more substitutions in human than in chimp. The accelerated elements, and in particular the top five, show a strong bias for adenine and thymine to guanine and cytosine nucleotide changes and are disproportionately located in high recombination and high guanine and cytosine content environments near telomeres, suggesting either biased gene conversion or isochore selection. In addition, there is some evidence of directional selection in the regions containing the two most accelerated regions. A combination of evolutionary forces has contributed to accelerated evolution of the fastest evolving elements in the human genome.
- A/T to G/C changes (weak to strong)
- located in high recombination areas
- next to genes that are enriched for transcription factors
- located in high G/C content areas near telomeres
Forces Shaping the Fastest Evolving Regions in the Human Genome
Katherine S. Pollard, Sofie R. Salama, Bryan King, Andrew D. Kern, Tim Dreszer, Sol Katzman1, Adam Siepel, Jakob S. Pedersen, Gill Bejerano, Robert Baertsch, Kate R. Rosenbloom, Jim Kent, David Haussler
PLoS Genetics vol. 2 issue 10; October 2006
Abstract: Comparative genomics allow us to search the human genome for segments that were extensively changed in the last ~5 million years since divergence from our common ancestor with chimpanzee, but are highly conserved in other species and thus are likely to be functional. We found 202 genomic elements that are highly conserved in vertebrates but show evidence of significantly accelerated substitution rates in human. These are mostly in non-coding DNA, often near genes associated with transcription and DNA binding. Resequencing confirmed that the five most accelerated elements are dramatically changed in human but not in other primates, with seven times more substitutions in human than in chimp. The accelerated elements, and in particular the top five, show a strong bias for adenine and thymine to guanine and cytosine nucleotide changes and are disproportionately located in high recombination and high guanine and cytosine content environments near telomeres, suggesting either biased gene conversion or isochore selection. In addition, there is some evidence of directional selection in the regions containing the two most accelerated regions. A combination of evolutionary forces has contributed to accelerated evolution of the fastest evolving elements in the human genome.
Tuesday, October 17, 2006
Back to blogging soon
I'm back from a short vacation and ready to get back to blogging... looking forward to the neanderthal sequence report coming out soon, supposedly
Monday, October 09, 2006
Bruce Lahn interview
Check out JP's post at Gene Expression where they have 10 questions for Bruce Lahn...some interesting tidbits, including his take on brain size difference among hominid species, how to test for signatures of selection, and about the future of research in human genetics with respect to niches for data miners vs. researhers who generate their own data.
Another way to estimate admixture
This looks like a STRUCTURE-type program.
Bayesian identification of admixture events using multilocus molecular markers.
Corander J, Marttinen P.
Molecular Ecology 2006 Sep;15(10):2833-43
Abstract: Bayesian statistical methods for the estimation of hidden genetic structure of populations have gained considerable popularity in the recent years. Utilizing molecular marker data, Bayesian mixture models attempt to identify a hidden population structure by clustering individuals into genetically divergent groups, whereas admixture models target at separating the ancestral sources of the alleles observed in different individuals. We discuss the difficulties involved in the simultaneous estimation of the number of ancestral populations and the levels of admixture in studied individuals' genomes. To resolve this issue, we introduce a computationally efficient method for the identification of admixture events in the population history. Our approach is illustrated by analyses of several challenging real and simulated data sets. The software (baps), implementing the methods introduced here, is freely available at http://www.rni.helsinki.fi/~jic/bapspage.html.
Bayesian identification of admixture events using multilocus molecular markers.
Corander J, Marttinen P.
Molecular Ecology 2006 Sep;15(10):2833-43
Abstract: Bayesian statistical methods for the estimation of hidden genetic structure of populations have gained considerable popularity in the recent years. Utilizing molecular marker data, Bayesian mixture models attempt to identify a hidden population structure by clustering individuals into genetically divergent groups, whereas admixture models target at separating the ancestral sources of the alleles observed in different individuals. We discuss the difficulties involved in the simultaneous estimation of the number of ancestral populations and the levels of admixture in studied individuals' genomes. To resolve this issue, we introduce a computationally efficient method for the identification of admixture events in the population history. Our approach is illustrated by analyses of several challenging real and simulated data sets. The software (baps), implementing the methods introduced here, is freely available at http://www.rni.helsinki.fi/~jic/bapspage.html.
Thursday, October 05, 2006
Do we know all the ways that humans got to the Americas?
A paper in Current Anthropology from a few months back that, using data on presence of hookworm the pre-Columbian hookworms and "paleoclimate modelling simulations" implies that there were other colonizers of the Americas, other than the Clovis ones who came through the Bering Strait. From the last paragraph:
The Americas might very well have been originally inhabited by humans moving along an interior land route across Beringia at around 13,000 years BP; however, our research indicates that the Clovis people were unlikely carriers of the hookworm, and this suggests that the parasites were introduced by some alternative mechanism. Alternative mechanisms include the possibility that the parasite entered the Americas by an interior route via Beringia during a period of lower sea level but with regional temperatures significantly higher than at 13,000 years BP. The archaeological literature offers a number of possible nonterrestrial routes, including transoceanic contacts between Japan and Central America (Meggers 1975) and coastal routes along the North Pacific (Heusser 1960; Fladmark 1979; Hetherington et al. 2003) and the North Atlantic (Bradley and Stanford 2004). Pointing to paleoparasitological findings, Araujo, Ferreira, and Confalonieri (1981; Araujo et al. 1988) have discussed the possibility of a trans-Pacific introduction.
Parasites, Paleoclimate, and the Peopling of the Americas
Using the Hookworm to Time the Clovis Migration
Alvaro Montenegro, Adauto Araujo, Michael Eby, Luiz Fernando Ferreira, Renée Hetherington, and Andrew J. Weaver
CURRENT ANTHROPOLOGY Volume 47, Number 1, February 2006
Abstract: Paleoparasitological findings and paleoclimate modelling simulations indicate that early peoples migrating via the "Clovis first" route across Beringia into North America could not have traversed the required distance in time to provide a reasonable explanation for the presence of the hookworm in the pre-Columbian Americas. The introduction of the hookworm into the Americas by a land migration at around 13,000 years BP could have happened only under extraordinary circumstances and even then would have required displacement rates that appear to have no parallel in the archaeology of the continent. This implies that while the Clovis people may have been the first migrants to the Americas, they were almost certainly not the only such migrants.
The Americas might very well have been originally inhabited by humans moving along an interior land route across Beringia at around 13,000 years BP; however, our research indicates that the Clovis people were unlikely carriers of the hookworm, and this suggests that the parasites were introduced by some alternative mechanism. Alternative mechanisms include the possibility that the parasite entered the Americas by an interior route via Beringia during a period of lower sea level but with regional temperatures significantly higher than at 13,000 years BP. The archaeological literature offers a number of possible nonterrestrial routes, including transoceanic contacts between Japan and Central America (Meggers 1975) and coastal routes along the North Pacific (Heusser 1960; Fladmark 1979; Hetherington et al. 2003) and the North Atlantic (Bradley and Stanford 2004). Pointing to paleoparasitological findings, Araujo, Ferreira, and Confalonieri (1981; Araujo et al. 1988) have discussed the possibility of a trans-Pacific introduction.
Parasites, Paleoclimate, and the Peopling of the Americas
Using the Hookworm to Time the Clovis Migration
Alvaro Montenegro, Adauto Araujo, Michael Eby, Luiz Fernando Ferreira, Renée Hetherington, and Andrew J. Weaver
CURRENT ANTHROPOLOGY Volume 47, Number 1, February 2006
Abstract: Paleoparasitological findings and paleoclimate modelling simulations indicate that early peoples migrating via the "Clovis first" route across Beringia into North America could not have traversed the required distance in time to provide a reasonable explanation for the presence of the hookworm in the pre-Columbian Americas. The introduction of the hookworm into the Americas by a land migration at around 13,000 years BP could have happened only under extraordinary circumstances and even then would have required displacement rates that appear to have no parallel in the archaeology of the continent. This implies that while the Clovis people may have been the first migrants to the Americas, they were almost certainly not the only such migrants.
Tuesday, October 03, 2006
A(nother) "muscle gene" shows population differences
 Dienekes reports on this in-press AJHG paper, by Matthew A. Saunders, Jeffrey M. Good, Elizabeth C. Lawrence, Robert E. Ferrell, Wen-Hsiung Li and Michael W. Nachman, entitled Human adaptive evolution at Myostatin, a regulator of muscle growth
Dienekes reports on this in-press AJHG paper, by Matthew A. Saunders, Jeffrey M. Good, Elizabeth C. Lawrence, Robert E. Ferrell, Wen-Hsiung Li and Michael W. Nachman, entitled Human adaptive evolution at Myostatin, a regulator of muscle growthThe authors find strong evidence of (maybe diversifying) selection in humans on some variants of the Myostatin gene (GDF8), which encodes a negative regulator of skeletal muscle growth. They also find somewhat large differences in frequency of these mutations between sub-Saharan Africans (up to 31%) and rare in others.
The authors don't mention much about the implications of this in terms of adaptation, except for a passing mention that "one of the many possible adaptive implications of such an effect could be protection from muscle-wasting in times of famine, a potentially recurrent phenomenon for early agricultural societies."
This gene falls in a line of other muscle related genes found to have population differences, the main one being ACTN3.
Monday, October 02, 2006
Review of programs for population genetics data analysis
This looks like a great paper. It is the first in a four paper focus on statistical genetics in the latest Nature Review Genetics. It describes and compares 20 or so programs based on what they can be used for, how they can be used, what type of data they can work with, their assumptions, gives links to their online resources, how the same data files can be used with several programs, computation time, etc... can make for great reference material.
Computer programs for population genetics data analysis: a survival guide
Laurent Excoffier and Gerald Heckel
Nature Reviews Genetics 7, 745-758 (October 2006)
Focus on: Statistical Analysis
Abstract: The analysis of genetic diversity within species is vital for understanding evolutionary processes at the population level and at the genomic level. A large quantity of data can now be produced at an unprecedented rate, requiring the use of dedicated computer programs to extract all embedded information. Several statistical packages have been recently developed, which offer a panel of standard and more sophisticated analyses. We describe here the functionalities, special features and assumptions of more than 20 such programs, indicate how they can interoperate, and discuss new directions that could lead to improved software and analyses.
Computer programs for population genetics data analysis: a survival guide
Laurent Excoffier and Gerald Heckel
Nature Reviews Genetics 7, 745-758 (October 2006)
Focus on: Statistical Analysis
Abstract: The analysis of genetic diversity within species is vital for understanding evolutionary processes at the population level and at the genomic level. A large quantity of data can now be produced at an unprecedented rate, requiring the use of dedicated computer programs to extract all embedded information. Several statistical packages have been recently developed, which offer a panel of standard and more sophisticated analyses. We describe here the functionalities, special features and assumptions of more than 20 such programs, indicate how they can interoperate, and discuss new directions that could lead to improved software and analyses.
Subscribe to:
Posts (Atom)

